[1]:
%pylab inline
Populating the interactive namespace from numpy and matplotlib
VASP - Surface energy
In this example, we introduce how to perform VASP calculations using strucscan and calculate the formation energy of an example (100) fcc surface in Ni. This example requires two prerequisites: * a licensed VASP version * a configured resource directory including the necessary POTCAR for Ni
In the documentation, it is explained how to set up the resource directory for VASP. We will also need a settings template. For this, you can stick to the default one that comes with the repository. We will perform a spin-polarised calculation with an energy cut-off of 500 eV:
[2]:
! cat ../resources/engines/vasp/settings/500_SP.incar
! ISIF and IBRION flags will be set automatically by strucscan
ALGO = Fast
PREC = Accurate
EDIFF = 1e-05
NSW = 100
NELM = 60
LREAL = .FALSE.
LWAVE = .FALSE.
ISPIN = 2
LCHARG = .FALSE.
LORBIT = 11
ENCUT = 500
The structure
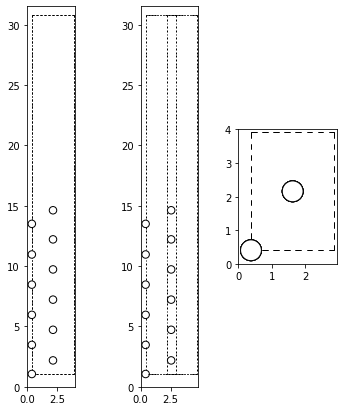
Let’s have a look at the surface structure that we want to investigate:
[3]:
from ase.visualize.plot import plot_atoms
from ase import io
structname = "../structures/unaries/surfaces/fcc_110surf_12at.cfg"
atoms = io.read(structname, format="cfg")
fig, axs = plt.subplots(1, 3, figsize=(6,7))
for ind, rotation in enumerate(['90x,90y', '90x,45y', '0x']):
plot_atoms(atoms, rotation=(rotation), ax=axs[ind])
plt.show()
The input dictionary
The next step is to set up the input dictionary properly. We can either start from scratch as in the previous example or start right away with the pre-implemented example for vasp:
[4]:
from strucscan.resources.inputyaml import *
input_dict = VASP().EXAMPLE
input_dict
[4]:
{'species': 'Ni Al_pv',
'engine': 'VASP 5.4',
'machine': 'example_vasp',
'ncores': '1',
'nnodes': '1',
'queuename': 'none',
'potential': 'PBE',
'properties': 'atomic',
'prototypes': 'L1_2',
'settings': '500_SP.incar',
'magnetic configuration': 'SP',
'initial magnetic moments': '2.0 0.',
'kdens': '0.15',
'kmesh': 'Monkhorst-pack',
'initial atvolume': 'default',
'verbose': False,
'monitor': True,
'submit': True,
'collect': False}
The example input is already configured for our example machine vasp_example which features no queueing system but we need to adapt some values:
[5]:
input_dict.update({"species": "Ni",
"potentials": "PBE",
"properties": "atomic",
"prototypes": "fcc.cfg fcc_110surf_12at.cfg",
"initial magnetic moments": "2.0",
"kdens": "0.15",
"verbose": True})
input_dict
[5]:
{'species': 'Ni',
'engine': 'VASP 5.4',
'machine': 'example_vasp',
'ncores': '1',
'nnodes': '1',
'queuename': 'none',
'potential': 'PBE',
'properties': 'atomic',
'prototypes': 'fcc.cfg fcc_110surf_12at.cfg',
'settings': '500_SP.incar',
'magnetic configuration': 'SP',
'initial magnetic moments': '2.0',
'kdens': '0.15',
'kmesh': 'Monkhorst-pack',
'initial atvolume': 'default',
'verbose': True,
'monitor': True,
'submit': True,
'collect': False,
'potentials': 'PBE'}
Please note that you may want to adapt the machine config.yaml to the machine on which you are running VASP. In this example, the config.yaml looks as following:
[6]:
from pprint import pprint
import yaml
with open("../resources/machineconfig/example_vasp/config.yaml", "r") as stream:
config = yaml.safe_load(stream)
pprint(config)
{'VASP': {'serial': 'module load vasp/5.4.4\nvasp_std\n'},
'scheduler': 'noqueue',
'smallest queue': None}
We also added fcc.cfg to our list of prototypes. This is the reference structure that we need to calculate the formation energy. Now, we can get started: ## Running strucscan
[ ]:
from strucscan.core.jobmanager import JobManager
JobManager(input_dict)
Data tree path: /home/users/pietki8q/git/strucscan-master/data
Structure repository: /home/users/pietki8q/git/strucscan-master/structures
Resource repository: /home/users/pietki8q/git/strucscan-master/resources
Optional key 'k points file' not provided. Default value will be used: (None)
key: : your input what strucscan reads
----------------------------------------------------------------------------------------------------
species : Ni Ni
engine : VASP 5.4 VASP 5.4
machine : example_vasp example_vasp
ncores : 1 1
nnodes : 1 1
queuename : none none
potential : PBE PBE
properties : atomic atomic
prototypes : fcc.cfg fcc_110surf_12at.cfg fcc.cfg fcc_110surf_12at.cfg
settings : 500_SP.incar 500_SP.incar
magnetic configuration : SP SP
initial magnetic moments : 2.0 2.0
kdens : 0.15 0.15
kmesh : Monkhorst-pack Monkhorst-pack
initial atvolume : default default
verbose : True True
monitor : True True
submit : True True
collect : False False
potentials : PBE PBE
k points file : (not set) (not set)
>> Initializing:
Initialized Ni atomic
Initialized Ni atomic
Initialized Ni atomic
2 jobs in JobList:
------------------------------------------------------------------------------------------------------------------
#: jobpath prototype path
------------------------------------------------------------------------------------------------------------------
0: VASP_5_4__500_kdens_0_150_SP_PBE/Ni/atomic__fcc_110surf_12at__Ni12 unaries/surfaces/fcc_110surf_12at.cfg
1: VASP_5_4__500_kdens_0_150_SP_PBE/Ni/atomic__fcc__Ni unaries/bulk/fcc.cfg
#: jobpath id status start end
------------------------------------------------------------------------------------------------------------------
0 VASP_5_4__500_kdens_0_150_SP_PBE/Ni/atomic__fcc_110surf_12at__Ni12 None does not exist
1 VASP_5_4__500_kdens_0_150_SP_PBE/Ni/atomic__fcc__Ni None does not exist
>> Entering loop:
Determining surface energy
After finishing successfully, strucscan saves the data from the calculation in dictionaries in a human-readable JSON format.
[ ]:
! ls ../data/VASP_5_4__500_kdens_0_150_SP_PBE/
[ ]:
import json
with open("../../VASP_5_4__500_kdens_0_150_SP_PBE__Ni__output_dict.yaml") as stream:
output_dict = json.load(stream)
stream.close()
from pprint import pprint
pprint(output_dict)
In order to calculate the surface energy with the following equation, we also need to determine the surface area A:
For this, we need to read the final surface structure manually.
[9]:
from ase import io
surface_atoms = io.read("../../data/VASP_5_4__500_kdens_0_150_SP_PBE/Ni/atomic__fcc_110surf_12at__Ni12/OUTCAR.gz", format="vasp-out")
surface_atoms
[9]:
Atoms(symbols='Ni12', pbc=True, cell=[1.978349761, 2.797335995, 23.740192654], calculator=SinglePointDFTCalculator(...))
[11]:
eV_A2_to_mJ_m2 = 1.60218e-19 * 1e20 * 1e3
surface_structure_energy = output_dict["atomic__fcc_110surf_12at__Ni12"]["structure_energy"]
surface_n_at = output_dict["atomic__fcc_110surf_12at__Ni12"]["n_atom"]
surface_area = np.linalg.det(surface_atoms.get_cell()[:2, :2])
basis_ref_energy = output_dict["atomic__fcc__Ni"]["structure_energy"]
basis_ref_n_atom = output_dict["atomic__fcc__Ni"]["n_atom"]
surface_formation_energy = (surface_structure_energy / surface_n_at - basis_ref_energy / basis_ref_n_atom) / (2 * surface_area)
print(surface_formation_energy * eV_A2_to_mJ_m2)
1379.5079416851156
[ ]: